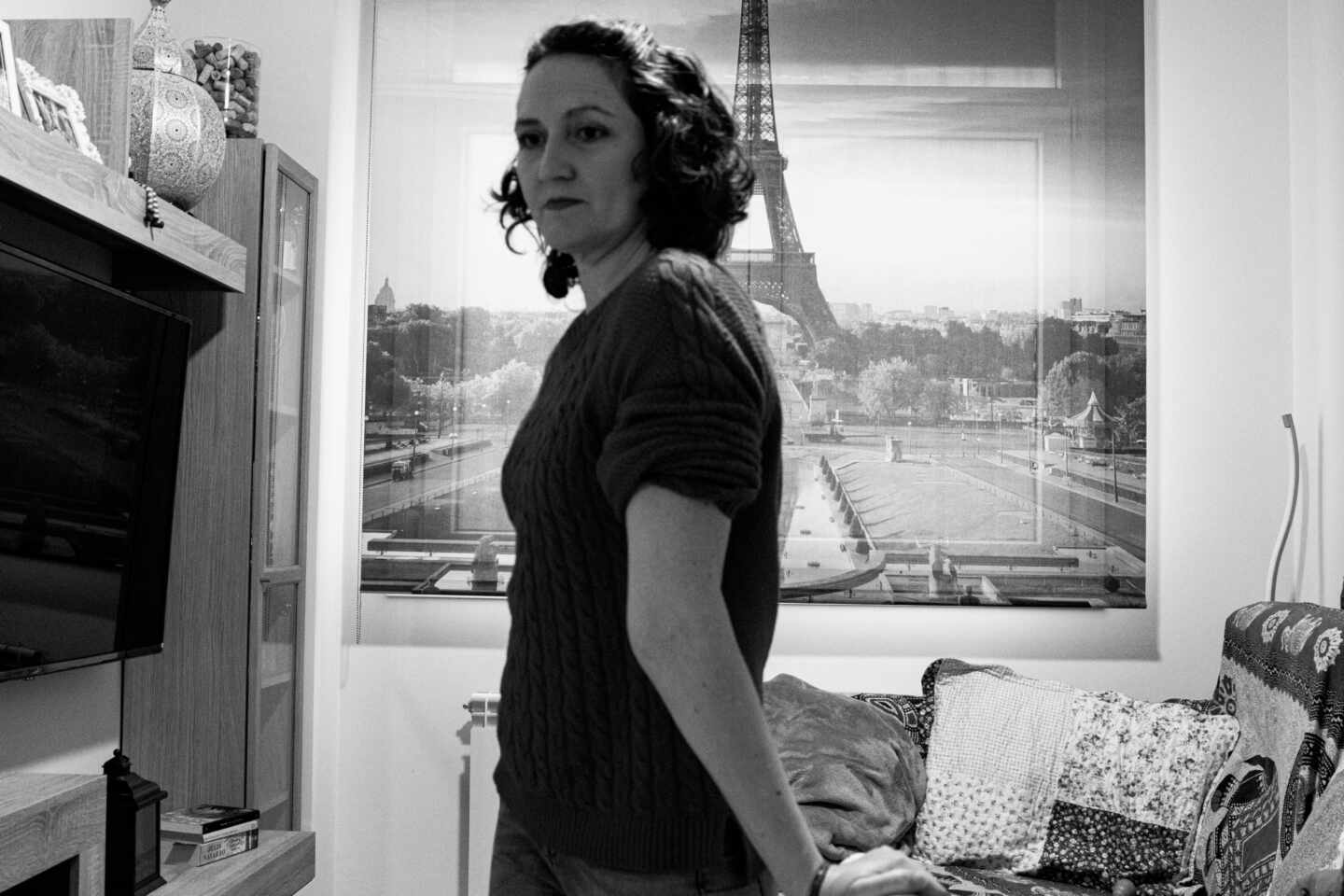
Si la media hasta el diagnóstico de una enfermedad rara son cinco años, quienes padecen el síndrome de Ehlers-Danlos no lo tienen hasta que cumplen entre 30 y 40 años. Elena Fernández tenía 32 cuando consiguió poner un nombre oficial a lo que le ocurría, aunque llevaba sufriendo los síntomas desde bien pequeña.
“A los cinco años ya me ponían inyecciones para el dolor en las articulaciones y decían a mis padres que tenía reuma”, recuerda Fernández, que padece esta enfermedad genética heredada de su madre y compartida con sus dos hermanos: “Mi madre la tenía pero no lo supo hasta después que yo. Mis hermanos también la padecen en distinta medida “.
El síndrome de Ehler Danlos (SED) es un conjunto de alteraciones genéticas cuyo denominador común es un defecto en el colágeno, cuya función es crear y mantener las estructuras de los tejidos del organismo. Quienes la padecen sufren múltiples alteraciones que pueden afectar a todos los órganos aunque uno de sus elementos más identificativos es la hiperlaxitud. “Somos personas cuyos movimientos naturales son de contorsionista y eso nos destroza los cartílagos. Nuestra piel también cicatriza mal y cualquier golpe se nos convierte en un gran moratón”, explica Fernández.
Pero no es solo la piel o los cartílagos. En algunos tipos de la enfermedad (hay 10 identificados y 23 mutaciones genéticas asociadas) lo más afectado son los vasos sanguíneos, cuya fragilidad hace que tiendan a romperse; en otros casos el tejido ocular, lo que eleva el riesgo de desprendimiento de retina. El intestino también suele sufrir de mala absorción.
Otro de los síntomas comunes a todos los enfermos es el gran dolor en las articulaciones, derivado de la hiperlaxitud. “El gran problema de esta enfermedad es que es difícil de ver por parte de los médicos. La laxitud produce síntomas en movimiento, pero no en estático y eso hace que, por ejemplo, un escáner no dé resultados anormales. Solo se ve en pruebas dinámicas en movimiento, y esas no se hacen en todos los hospitales”, indica Bárbara Otero, médica internista del Hospital 12 de Octubre de Madrid y responsable del área de colagenopatías hereditarias de la Unidad de Enfermedades Minoritarias.
Una infancia "más o menos normal"
“Mi infancia fue más o menos normal, debido a los problemas intestinales vomitaba mucho, pero decían que yo era una niña delicada o nerviosa. Nadie sabía lo que pasaba. Yo era muy deportista pero a los 15 años me quedé sin cartílago en la rodilla y sufrí la primera operación”, explica Fernández.
Desde entonces, esta madrileña de 41 años se ha operado cinco veces. La rodilla, dos hernias discales, la cadera y una intervención por un embarazo ectópico. Es muy difícil que alguien con SED pueda quedarse embarazada ya que hay una difícil implantación y el útero tiende a desprenderse.
Yo sufría muchos dolores pero los médicos no veían el origen y me derivaban al psicólogo
Tras esa primera operación de rodilla, se agudizaron los dolores y con ellos el periplo de médicos y psicólogos que atendían sus síntomas de manera deslavazada. “Yo sufría muchos dolores pero los médicos no veían el origen y me derivaban al psicólogo”, relata Fernández, para quien “lo peor de la enfermedad no ha sido la propia enfermedad sino la incomprensión por parte de la sociedad”.
Ese es el punto en común en casi todos los que padecen este síndrome, “la incomprensión, el desconocimiento y que prácticamente las tildan de locas, porque la mayoría son mujeres”, reconoce Otero. Se estima que alrededor de una por cada 5.000 personas padecen esta enfermedad y eso la encuadra entre las enfermedades raras, que este 29 de febrero celebran su día mundial.
Que Otero trate ahora a Fernández es el resultado de un viaje que duró más de dos décadas décadas. Hasta los 22 años esta mujer no escuchó “síndrome de Ehlers Danlos”. “Me lo dijo un médico privado que estaba tratando a mi madre. Fui por dolor en el cuello y me miró más allá, observó mis pies. Me dijo: tienes el síndrome de Ehlers Danlos y tu médico de cabecera a partir de ahora tiene que ser un reumatólogo”. Ojalá. Porque aquel fue el inicio de la peor parte del viaje de Fernández.
“Yo soy muy positiva. Pero todo lo que he sufrido por la enfermedad es menos de lo que me ha hecho la incomprensión, el aislamiento de la sociedad”, recalca Fernández, que tras escuchar aquellas palabras inició una lucha por conseguir un examen genético que confirmara el diagnóstico.

"Para qué quieres saber cuál es tu enfermedad"
De reumatólogo en reumatólogo, por consultas en las que tenía que deletrear el síndrome al médico, aún pasaron seis años hasta que con 28 consiguió que le realizaran el test genético en el Hospital de la Princesa. Y se lo hicieron mal. “Me analizaron otro tipo de la enfermedad y salió negativo. Sabía que era otro tipo porque el mío se caracteriza por otros síntomas. Pero me dijeron que el hospital no aprueba la realización de otra prueba por su coste. Tuve que escuchar a la genetista diciéndome que para qué quería saber cuál era mi enfermedad, si era degenerativa y no tiene cura…”
Palo tras palo, Fernández no se daba por vencida. Llamó a todas las puertas y se resistía a tocar fondo cuando una amiga traductora la animó a buscar soluciones fuera de España. “Encontramos un especialista en Bélgica y me compré un billete. Fui con mi madre y mi amiga y en cinco minutos me dijo lo que tenía. Me hicieron las pruebas sobre la marcha y al fin pude conseguir mi diagnóstico.
El diagnóstico, "paz interior"
Fernández define ese momento como “un descanso, una gran paz interior, poder callar muchas bocas”. Porque hasta entonces la enfermedad se había cobrado mucho aparte de su salud. Había perdido el trabajo y con tesón sacó una oposición “en nueve meses y con un corsé desde la frente al coxis”. También perdió parejas y la posibilidad de tener un hijo cuando vio que podía costarle la vida. “Yo soy súper positiva pero no es lo normal en mi enfermedad. Suele ser una enfermedad depresiva porque cuando arreglas una cosa se rompe otra… Es todo el cuerpo y eso mina. Cuando una persona está enferma necesita comprensión, no críticas”.
La falta de conocimientos hace que esta enfermedad esté muy infradiagnosticada
Aunque en la mayoría de los casos de SED la esperanza de vida no está mermada, sí su calidad. "Tienen muchos problemas en su vida. Pierden el trabajo por la acumulación de bajas, no pueden hacer las actividades normales de cualquier persona y en casos severos pueden acabar en silla de ruedas", explica Otero. Los dolores y la situación de Fernández, por ejemplo, le confieren una discapacidad reconocida del 57%. Por el SED y por otra enfermedad rara que la primera le ha provocado, disautonomía, derivada de los problemas en las venas y que afecta a su riego sanguíneo.
Sin tratamientos disponibles "y muy poca investigación", reconoce Otero, lo que pueden los pacientes de SED es "prevenir cuidando la musculatura y tratar los síntomas como el dolor". La falta de conocimientos hace que esta enfermedad esté muy infradiagnosticada. La situación va mejorando pero los pacientes que llegan a mi unidad han pasado por un largo periplo de médicos", reconoce la internista.
Esta incomprensión o al menos la falta de un diagnóstico y un tratamiento certeros es algo que acusan casi la mitad de quienes padecen una enfermedad rara, según los datos de Feder. El 46,6% se queja de la atención sanitaria que recibe y el 72% dice haber recibido, al menos una vez, un tratamiento adecuado.
Sin tratamientos, Fernández se alegra de que actualmente es tratada por una médica que conoce bien su enfermedad. Su optimismo es admirable y contagioso: "No me lamento porque gracias a la enfermedad soy quien soy", afirma.
Te puede interesar




Lo más visto